Questa è una spiegazione dei test genetici disponibili per le cause note della sindrome di Angelman. È scritto per i genitori e per i non addetti ai lavori che non hanno una conoscenza pratica della genetica o della biologia molecolare, ma che vorrebbero capire i dettagli più tecnici di questi test.
(a cura della dott.ssa Ornella Rondinone, Specilizzanda in Genetica Medica, Università degli Studi di Milano)
La diagnosi della sindrome di Angelman (AS) si basa essenzialmente sulle caratteristiche cliniche del paziente, e/o sui risultati ottenuti da test genetici molecolari che suggeriscono mancanza o funzione insufficiente dell’allele ereditato dalla madre del gene UBE3A, localizzato nel cromosoma 15.
Benché nella maggior parte dei casi la diagnosi è clinica, la conferma molecolare è fondamentale per evidenziare il tipo specifico di alterazione molecolare perchè offre importanti elementi aggiuntivi, il primo è la possibilità di individuare il possibile rischio di ricorrenza nella famiglia.
La malattia insorge a seguito di alterazioni di una regione del cromosoma 15 (15q11.2-q13) ereditato dalla madre, dove mappa un gruppo di geni la cui espressione è monoallelica, diversamente dalla maggior parte dei geni che mostrano espressione biallelica. I geni implicati sono infatti classificati, sulla base della loro espressione, come soggetti a imprinting genomico, fenomeno che, attraverso modificazioni del DNA (principalmente la metilazione di siti specifici), determina l’espressione monoallelica di specifici geni. Significa che solo uno dei due alleli si esprime, a secondo della sua derivazione parentale. Per il gene causativo della AS si esprime solo l’allele materno, per altri geni soggetti ad imprinting l’espressione può avvenire anche esclusivamente per l’allele paterno (Figura 1).
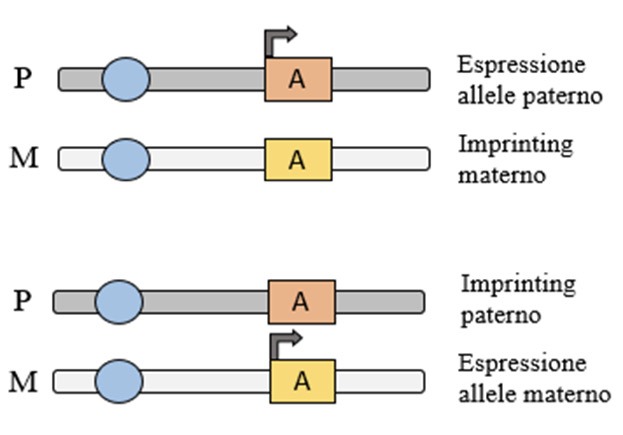
Figura 1. Imprinting genomico. Espressione monoallelica (solo espressione paterna o solo espressione materna) del gene A soggetto ad imprinting. Nella sindrome di Angelman l’espressione è solo materna.
Il gene implicato nella patogenesi della AS è UBE3A e i difetti genetici nella sindrome sono molteplici e tutti determinano mancata o carente funzione del gene (Fig. 2). Nello specifico, la causa più frequente (70%) è la microdelezione della regione 15q11.2-q13 di derivazione materna, mentre molto più rara è la disomia uniparentale paterna (UPD). In altri casi (2-3%), la sindrome è dovuta a difetti primari nel centro dell’imprinting (ICD) con perdita di metilazione del DNA. Mutazioni sull’allele materno del gene UBE3A, infine, sono causative della malattia nel 5-7% dei pazienti.

Figura 2. Meccanismi genetici causativi di AS
Per evidenziare la lesione genetica della AS è necessario procedere con più approcci molecolari, proprio per cercare di evidenziare l’alterazione nel singolo paziente (Tab 1). Nella Fig. 3 è indicato uno schema del percorso diagnostico. Va sottolineato che la sindrome può essere causata non solo da errori della sequenza del DNA che coinvolgono la regione del cromosoma 15 implicata, ma anche da difetti (delezioni, UPD, difetti di epigenetici di metilazione del centro dell’imprinting) che causano tutti anomala metilazione della regione cromosomica implicata. In generale, l’epigenetica consiste in una serie di modificazioni chimiche del DNA e della cromatina (DNA complessato con proteine a formare poi i cromosomi) che hanno il ruolo molto importante di far sì che i geni siano attivi o silenziati. UBE3A è soggetto a imprinting grazie ai meccanismi epigenetici che spengono (silenziano), in ognuno di noi l’allele che abbiamo ereditato padre, e fa rimanere acceso, quindi attivo quello di derivazione materna. Il principale sistema che le cellule usano per silenziare un gene è la metilazione del DNA in regioni regolatorie specifiche per ogni gene.
Misurando la metilazione nella regione specifica di regolazione dell’imprinting di UBE3A possiamo, già nell’ l’80% dei casi riscontrare una alterazione negli individui affetti, compresi quelli con delezione, UPD o ICD e le rare (meno dell’1%) condizioni di riarrangiamento cromosomico citogeneticamente visibile (cioè traslocazione o inversione). L’analisi della sequenza UBE3A rileva invece varianti patogenetiche in un ulteriore 11% circa degli individui che causano la assenza della proteina codificata da UBE3A o la sua scarsa funzione. Pertanto, i test genetici molecolari (analisi di metilazione e analisi di sequenza UBE3A) identificano alterazioni in circa il 90% degli individui. Esiste una piccola porzione di individui con caratteristiche fenotipiche classiche di AS in cui la diagnosi molecolare non identifica alterazioni.
Analogamente ad altre condizioni genetica a causa nota, i più recenti avanzamenti tecnologi permettono oggi la caratterizzazione di lesioni genetiche e genomiche non rilevabili con approcci quali l’analisi del cariotipo classico.

Figura 3. Percorso diagnostico della sindrome di Angelman
Approccio citogenetico
L’analisi citogenetica standard permette di evidenziare cambiamenti dell’assetto cromosomico, come delezioni, duplicazioni o riarrangiamenti strutturali che coinvolgono ampie porzioni cromosomiche. Per questo, il cariotipo è in grado di diagnosticare solo ampie delezioni, traslocazione o inversioni della regione 15q11.2-q13, errori genetici che si osservano complessivamente in meno dell’1% dei pazienti. La più comune delezione di 5-7 Mb (mega basi di sequenza) non è generalmente rilevata dall’analisi cromosomica di routine.
Test molecolari
Il test di metilazione della regione 15q11.2-q13, eseguito mediante MS-PCR (Methylation Specific PCR), pirosequenziamento e, più recentemente, mediante MS-MLPA (Methylation Specific Multiplex Ligation – Dependent Probe Assay), permette di rilevare difetti di metilazioni ascrivibili a microdelezione, disomia uniparentale (UPD) o difetti nel centro dell’imprinting (ICD), confermando la diagnosi in circa l’80% dei pazienti (Fig 4).
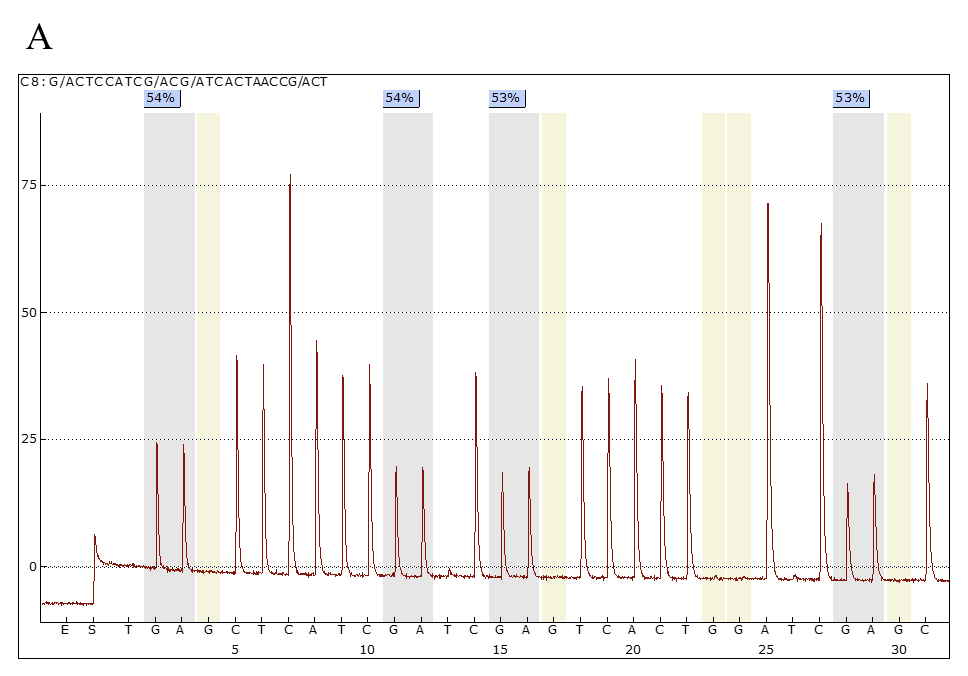
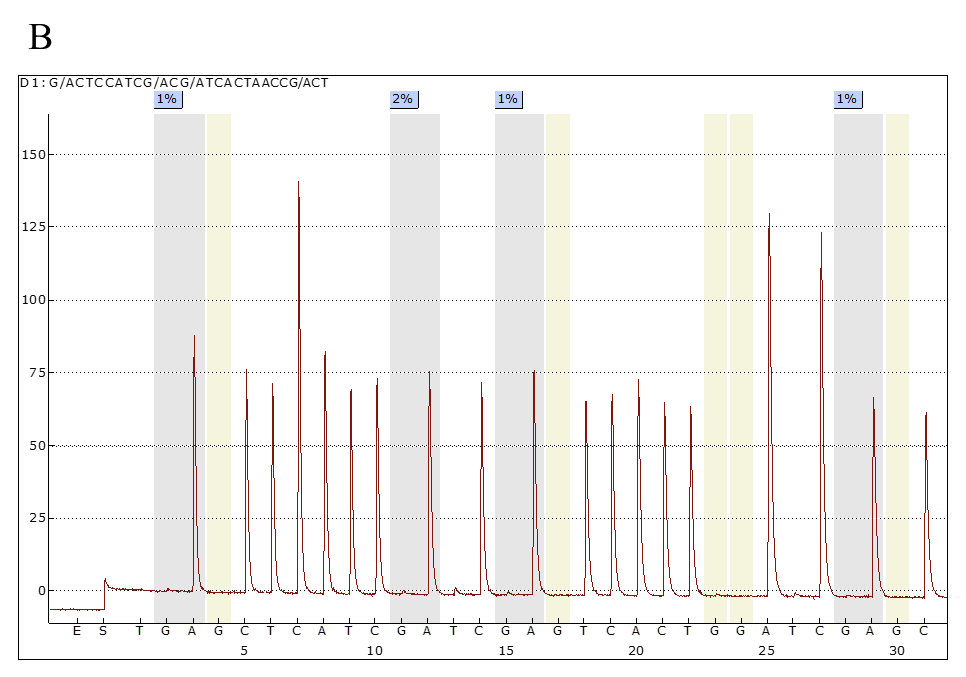
Figura 4. Pirosequenziamento per la valutazione dello stato di metilazione dei siti CpG nella regione regolatoria del cromosoma15q11.2-q13. A) Controllo sano: profilo di metilazione normale (media del 50%) dovuto alla presenza sia dell’allele materno (metilato) che dell’allele paterno (non metilato); B) Paziente AS: profilo di metilazione caratterizzato dall’assenza dell’allele materno (metilato), con effetto finale di ipometilazione del locus (1-2%). Le regioni in grigio rappresentano i siti di metilazione indagati, in azzurro la percentuale della loro metilazione.
Il test di ibridazione fluorescente in situ (FISH), effettuato con la sonda UBE3A/D15S10 e/o SNRPN, rileva la presenza di una delezione di circa 4-6 Mb nella regione 15q11.2-q13, presente in circa il 70% dei pazienti AS (Fig. 5). Il microarray cromosomico (CMA) può ulteriormente affinare la dimensione della delezione, che è stata dimostrato essere correlata alla gravità del fenotipo clinico (Fig 6).

Figura 5. Analisi in FISH con sonda specifica per evidenziare delezione del cromosoma 15. La freccia indica il cromosoma 15 in cui manca la porzione con segnali di ibridazione presenti invece nel cromosoma omologo normale (segnali arancio centrali).
Per approfondimenti:
GeneReviews. https://www.ncbi.nlm.nih.gov/books/NBK1144/
OMIM: https://www.omim.org/entry/105830?search=angelman&highlight=angelman
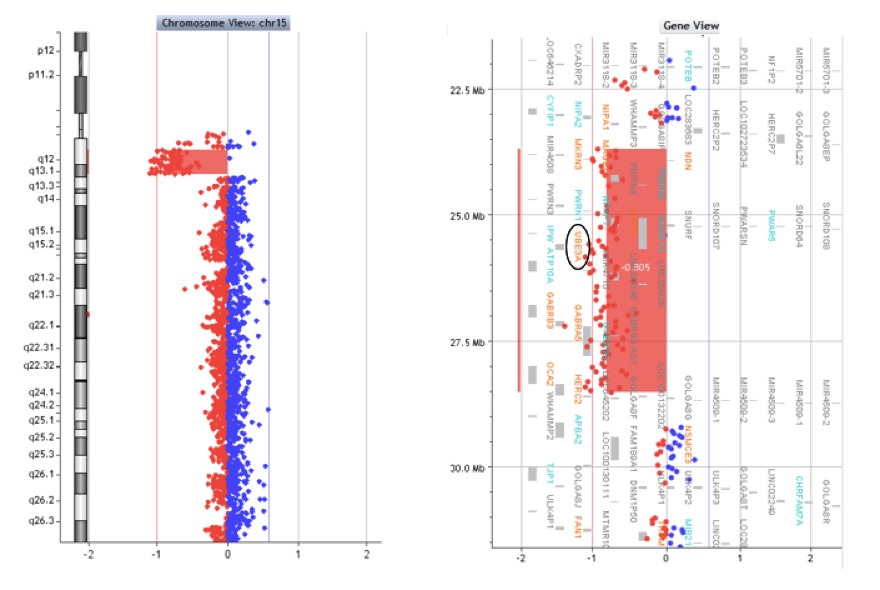
Figura 6. Analisi con microarray cromosomici (CMA). Microdelezione del braccio lungo del cromosoma 15 (del15q) materno nella regione 15q11.2-q13. La regione deleta è evidenziata dei segnali rossi a sinistra rispetto ai valori normali posti vicino al valore 0.Tra i geni coinvolti nella delezione vi è il gene UBE3A (cerchiato in nero nell’immagine a destra), la cui assenza di espressione causa la sindrome di Angelman.
Se un individuo risulta positivo sia al test di metilazione sia all’analisi FISH, possiamo confermare che il soggetto ha l’AS causata da delezione. Se un individuo risulta, invece, positivo al test di metilazione, ma il test FISH è negativo, avrà probabilmente UPD o ICD. L’analisi di marcatori molecolari verrà, quindi, utilizzata per determinare se l’individuo ha due copie del cromosoma 15 del padre (UPD) o se l’individuo ha un cromosoma 15 ereditato da ciascun genitore, ma con metilazione errata (ICD). In quest’ultimo caso, anche se l’individuo ha ereditato una copia del cromosoma 15 dalla madre e, quindi, il gene UBE3A è presente, il pattern di metilazione stabilito non è corretto e il gene di origine materna è silenziato.
Circa il 20% dei soggetti ha un quadro clinico caratteristico dell’AS, ma risulta negativo ai test molecolari elencati precedentemente. In tal caso è necessario ricorrere all’analisi di sequenza direttamente del gene UBE3A e capire se questo cambiamento corrisponde ad una mutazione patogenetica posta sull’allele del gene ereditato dalla madre.
Consulenza genetica
Il rischio di ricorrenza di sindrome di Angelman nei fratelli di un soggetto affetto dipende dal meccanismo genetico che determina la perdita di funzione del gene UBE3A. In genere, il rischio è meno dell’1% nel caso di probandi affetti per delezione della copia materna della regione 15q11.2-q13 o per UPD paterna. Il rischio di ricorrenza arriva fino al 50% per i casi in cui la sindrome è dovuta a mutazioni ereditate della madre del gene UBE3A. I riarrangiamenti cromosomici, invece, possono essere ereditati, anche se spesso si presentano come condizioni de novo e, quindi, con rischio di ricorrenza trascurabile. La diagnosi prenatale per gravidanze a rischio aumentato viene considerata quando il meccanismo genetico causativo è noto.
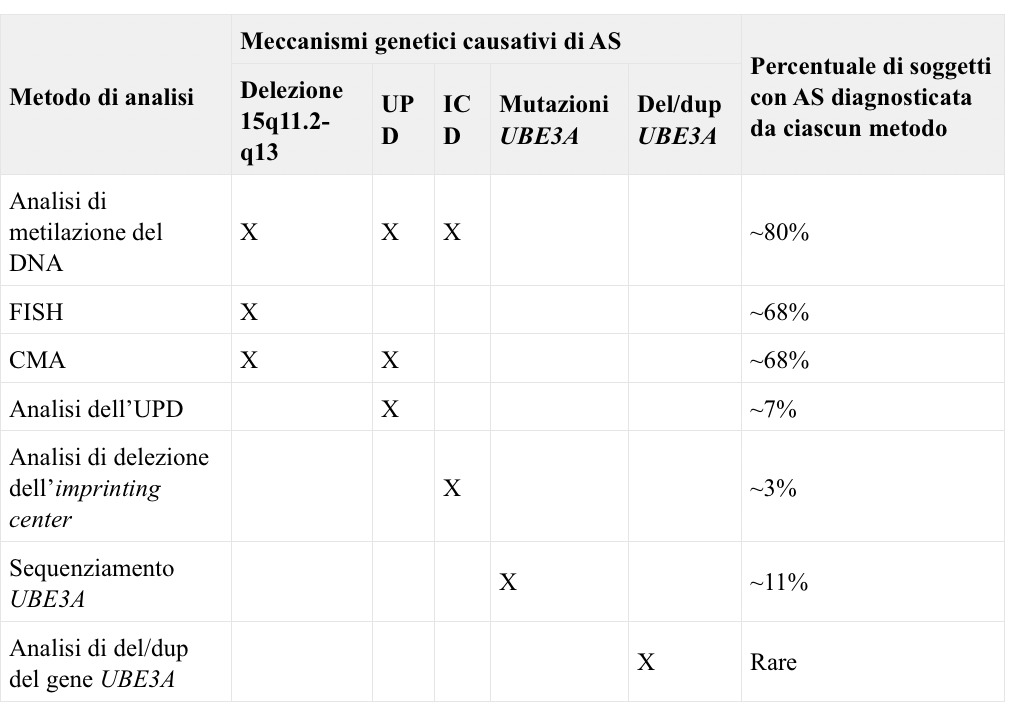
Tabella 1. Approcci molecolari utili per la diagnosi di AS e relativa percentuale di casi (%) diagnosticata da ciascuna metodica.
FAST si impegna ad aiutare le persone che vivono con la sindrome di Angelman a realizzare il loro pieno potenziale e migliorare la qualità della vita.
Link veloci…
Restiamo in contatto
- Sede legale Via Carioni 26, 20064, Gorgonzola(MI)
- Sedi operative:
- Viale Margherita di Savoia 1, 20900, Monza(MB)
- Via Rossini 27, 25010 San Zeno Naviglio (BS)
- Via Mansueto n.55/14, 16159 Genova
- Codice fiscale: 91584170152
- E-mail: info@CureAngelman.it
